Published in IJCP
August 2023
Case Report
Laurence-Moon-Bardet-Biedl Syndrome: A Rare Case Report
August 10, 2023 | Mahesh Dave, Anuj Goyal, Deepanshu, Ram Gopal Saini, Gaurav Dave
Family Medicine
https://doi.org/10.59793/ijcp.v34i3.574
Abstract
Introduction: Laurence-Moon-Bardet-Biedl syndrome (LMBBS) is a rare autosomal-recessive disorder, which is characterized principally by cardinal symptoms of marked central obesity, retinal dystrophy, polydactyly, mental retardation, hypogonadism and renal dysfunction. Case presentation: We report a case of a 20-year-old male who presented to us with history of fever, vomiting and dizziness. He was incidentally diagnosed as a case of LMBBS as the patient was having polydactyly, retinitis pigmentosa, central obesity, hypogonadism and low IQ. Conclusion: Laurence-Moon-Bardet-Biedl syndrome is a very rare syndrome with very low incidence; hence, we are reporting this case. In addition, we advice more diligent approach from various specialties, so that this syndrome can be picked up at an early age.
Keywords: Laurence-Moon-Bardet-Biedl syndrome, polydactyly, hypogonadism
The Laurence-Moon-Bardet-Biedl syndrome (LMBBS) is a rare autosomal recessive disorder which is characterized by symptoms of obesity, polydactyly, retinitis pigmentosa, hypogonadism, mental retardation and renal dysfunction.1 The frequency of the syndrome is estimated to be 1:1,60,000.2 Less than 15 cases have been reported from India.3 LMBBS may also present with other secondary abnormalities, including speech disorder and developmental delay, ataxia, diabetes insipidus and dental crowding.4 Due to involvement of photoreceptors and macula in eyes, it leads to night blindness and pigmentary retinopathy finally leading to complete loss of vision in later part of disease.5 Truncal obesity with normal birth weight is present in archetypal patients.6 Postaxial polydactyly with extra digit in hand or toe is present. Further, hypogonadism may manifest either as small testes with micropenis in males or underdeveloped uterus and fallopian tubes with menstrual irregularities in females. Therefore, LMBBS is a multiorgan involvement syndrome with rare occurrence and uneven life span.4 Hence, we hereby present a rare case of LMBBS.
CASE PRESENTATION
A 20-year-old male was patient admitted in medical ward, RNT Medical College, Udaipur, Rajasthan (India) with chief complaints of high-grade fever, vomiting and dizziness. On general physical examination, he was found to be overweight with central obesity. Incidentally, he was found to have 6 digits in all the four limbs (Fig. 1). On further enquiry, patient’s mother gave a history of diminished vision from both the eyes since early childhood which was progressive in nature. They had consulted an ophthalmologist who diagnosed it as retinitis pigmentosa for which he was on symptomatic treatment without any improvement. Further, his parents gave history of mental retardation with low IQ and happy and cheerful behavior. On further evaluation, he was found to have micropenis with bilateral testicular atrophy with decreased secondary sexual characters. His height was 170 cm and weight 79 kg with a body mass index (BMI) of 27.33 (pre-obese).
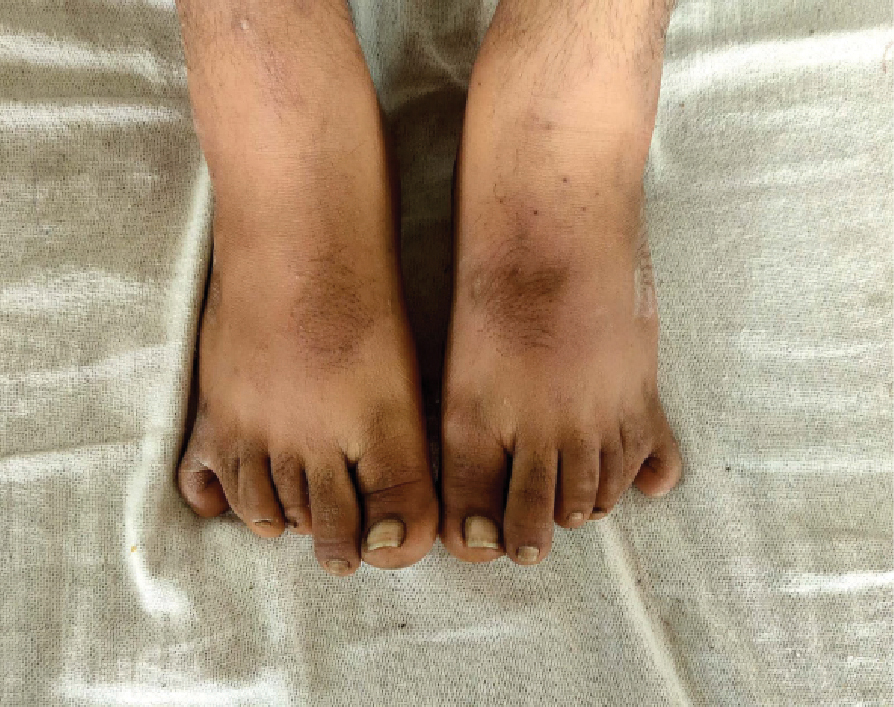
Figure 1. Showing hexadactyly in both lower limbs.
After history and clinical examination, we put our diagnosis as acute febrile illness with retinitis pigmentosa, central obesity, polydactyly and hypogonadism clinically suspected to be LMBBS.
Patient was investigated extensively to confirm our clinical diagnosis. Complete hemogram revealed normal hemoglobin levels, normal total leukocyte count, with mild thrombocytopenia (10,0000/mm3). Liver function tests showed normal bilirubin with mildly increased serum aminotransferase levels (SGOT 94 U/L and SGPT 80 U/L). Renal function tests were within normal limits. Urinalysis revealed proteinuria and glycosuria. Chest X-ray and ECG were normal. His fever profile revealed Plasmodium falciparum malaria. Ultrasonography showed mild hepatosplenomegaly with bilateral renal parenchymal Grade 1 disease with decreased testicular volume.
Specific investigations were done thereafter. Follicle-stimulating hormone (FSH) and luteinizing hormone (LH)
and testosterone levels were within normal limit but his prolactin levels were raised (400.6 uIU/mL). 2D-echocardiography was normal. Fundus examination revealed waxy pallor of disc, vessels attenuation and bony spicules in bilateral eyes (Fig. 2), which confirms the diagnosis of retinitis pigmentosa. Pure-tone audiometry revealed bilateral mild hearing loss (Fig. 3).

Figure 2. Fundus showing arterial attenuation, pale waxy disc with bony spicules in mid periphery.
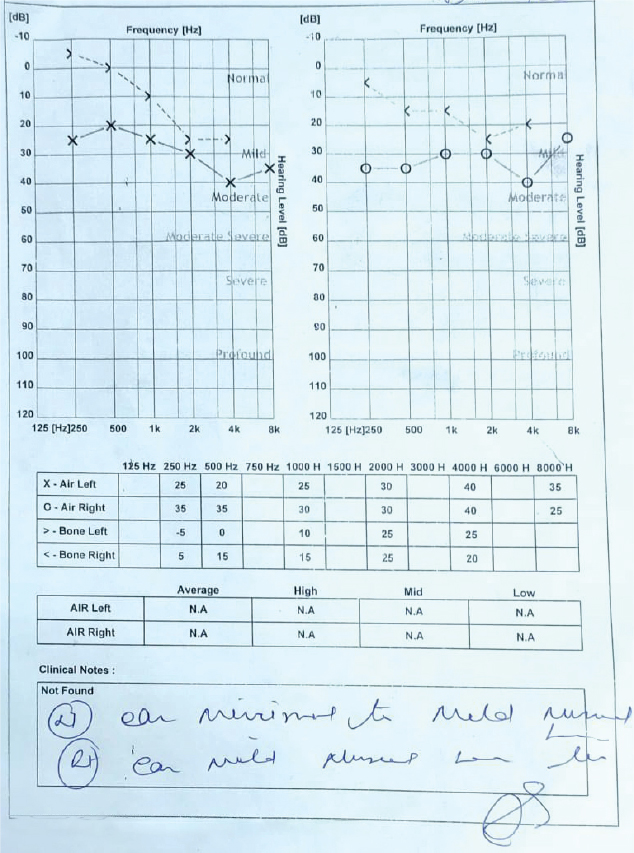
Figure 3. Pure-tone audiometry showing minimal to mild hearing loss in both the ears.
Hence, we confirm our diagnosis of LMBBS. The patient was treated symptomatically and discharged and was advised regular follow-up.
DISCUSSION
The LMBBS is an idiosyncratic, heterogeneous autosomal-recessive disorder with various clinical features requiring a multidisciplinary approach for early diagnosis. Literature has revealed that Laurence-Moon syndrome (LMS) and Bardet-Biedl syndrome (BBS) are two distinct disorders with overlapping features and very minute distinguishing elements between them.7 Most of the studies suggests that polydactyly and obesity is commonly observed in BBS, whereas spasticity is mainly seen in LMS but both of them can be considered same as LMBBS.8 So, we regarded our patient as LMBBS–a globally acquired label to elucidate the illness.9 Apart from the cardinal manifestations, other features of the BBS include various degrees of intellectual impairment, congenital heart block, brachycephaly, deafness and dental anomalies. The full spectrum of clinical features is found in only 40% to 45% of LMBBS cases.10 The detailed biochemical mechanism that leads to BBS is still unclear. Twelve genes (BBS1 to BBS12) that are responsible for the disease have been cloned. The BBS proteins are components of the centrosome and affect the ciliary transport; hence, the disease falls under the spectrum of "ciliopathies".11 In order to confirm the disease based on the clinical foundation, there is a revised criterion with certain primary/major features and secondary/minor features which are described in Tables 1 and 2.12 For diagnosis of LMBBS, it requires 4 major or 3 major together with 2 minor traits to confirm the diagnosis as suggested by Forsythe and Beales.13 The chief complaint includes night blindness with photophobia and visual blurring.5 Truncal obesity, sometimes with diabetes mellitus is a prominent feature.5 Polydactyly is the most phenotypically evident abnormality with a prevalence of 69%.14 Hypogonadism is the final primary symptom with a prevalence of 59%, which may be diagnosed during the stage of puberty with delayed secondary sexual characteristics. This syndrome requires early diagnosis and a multidisciplinary approach for quality management. Treatment options are limited either to conservative therapy or surgical management as per the symptoms. Spectacles with visual aids can improve visual quality.15 Physical activity with low-calorie and high-protein diet can be helpful in obesity.16 Polydactyly can be treated surgically while testosterone supplementation can be given to combat hypogonadism. Further, regular follow-ups with endocrine monitoring should be done. Educational and behavioral assessment should be done by clinical psychologist. Family members counseling sessions to be done for better patient care. Community needs to welcome these patients and not treat these patients with hatred and inequity. This can help in upgrading their status of living.
|
Table 1. Modified Diagnostic Criteria for LMBBS Representing Primary Features
|
|
Primary features
|
Present (+) or absent (-)
|
|
Rod-cone dystrophy
|
+
|
|
Polydactyly
|
+
|
|
Obesity
|
+
|
|
Learning disabilities
|
+
|
|
Hypogonadism in males
|
+
|
|
Renal anomalies
|
+
|
|
Table 2. Modified Diagnostic Criteria for LMBBS Representing Secondary Features
|
|
Secondary features
|
Present (+) or absent (-)
|
|
Speech disorder/delay
|
+
|
|
Strabismus/cataract/astigmatism
|
+
|
|
Brachydactyly/syndactyly
|
+
|
|
Developmental delay
|
+
|
|
Polyuria/polydipsia
|
-
|
|
Ataxia/poor coordination/imbalance
|
-
|
|
Mild spasticity
|
-
|
|
Diabetes mellitus
|
-
|
|
Dental crowding/hypodontia/small roots/high arched palate
|
-
|
|
Left ventricular hypertrophy/Congenital heart diseases
|
-
|
|
Hepatic fibrosis
|
-
|
CONCLUSION
Laurence-Moon-Bardet-Biedl syndrome is rare entity with very low incidence. Even though the patient was seen by different specialists earlier, the diagnosis was missed due to the rarity of this condition. This case report highlights the features required to diagnose this syndrome and will help in diagnosis of this syndrome at an early age.
REFERENCES
- Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999;36(6):437-46.
- Klein D, Ammann F. The syndrome of Laurence-Moon- Bardet-Biedl and allied diseases in Switzerland. Clinical, genetic and epidemiological studies. J Neurol Sci. 1969;9(3):479-513.
- Hooda AK, Karan SC, Bishnoi JS, Nandwani A, Sinha T. Renal transplant in a child with Bardet-Biedl syndrome: a rare cause of end-stage renal disease. Indian J Nephrol. 2009;19(3):112-4.
- Forsyth R, Gunay-Aygun M. Bardet-Biedl syndrome overview. 2003 Jul 14 [updated 2020 Jul 23]. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, et al (Eds.). GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023.
- Green JS, Parfrey PS, Harnett JD, Farid NR, Cramer BC, Johnson G, et al. The cardinal manifestations of Bardet-Biedl syndrome, a form of Laurence-Moon-Biedl syndrome. N Engl J Med. 1989;321(15):1002-9.
- Anosov M, Birk R. Bardet-Biedl syndrome obesity: BBS4 regulates cellular ER stress in early adipogenesis. Mol Genet Metab. 2019;126(4):495-503.
- Ehsan U, Mudassar I. Laurence Moon Biedl syndrome in a young girl with overlapping features of Turner syndrome: a case report. Med Channel. 2010;16(3):465-7.
- Moore SJ, Green JS, Fan Y, Bhogal AK, Dicks E, Fernandez BA, et al. Clinical and genetic epidemiology of Bardet-Biedl syndrome in Newfoundland: a 22-year prospective, population-based, cohort study. Am J Med Genet A. 2005;132A(4):352-60.
- Khan OA, Majeed R, Saad M, Khan A, Ghassan A. Rarity of Laurence Moon Bardet Biedl syndrome and its poor management in the Pakistani population. Cureus. 2019;11(2):e4114.
- Prosperi L, Cordella M, Bernasconi S. Electroretinography and diagnosis of the Laurence-Moon-Bardet-Biedl syndrome in childhood. J Pediatr Ophthalmol. 1977;14(5):305-8.
- Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peränen J, Merdes A, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129(6):1201-13.
- Mahmood SH, Khan M, Qadar LT, Yousuf F, Hasan M. A unique manifestation of Bardet-Biedl syndrome with otolaryngologic symptoms and bronchopneumonia in a one-year-old girl. Cureus. 2019;11(9):e5717.
- Forsythe E, Beales P. Bardet-Biedl syndrome. Eur J Hum Genet. 2013;21(1):8-13.
- Qadar LT, Ahmed ZM, Munawar M, Hasan CA, Iqbal SU. Laurence-Moon-Bardet-Biedl syndrome with coexisting abdominal distension and positive fluid thrill: a rare manifestation reported in Karachi, Pakistan. Cureus. 2019;11(6):e4885.
- Khan PA, Nishaat J, Noor S, Fatima N. Laurence Moon Bardet Biedl syndrome: a rare case report in a tertiary care teaching hospital, Hyderabad, Telangana, India. Int J Med Public Health. 2017;7(1):68-71.
- Khan BA, Shahid A, Bin Nazir M, Khan KS, Punshi A. Laurence-Moon-Bardet-Biedl syndrome: a case report. Cureus. 2019;11(9):e5618.
|