Abstract
Catecholamine-secreting tumors occur with equal frequency in men and women, primarily in the fourth and fifth decades. The associated hypertension may be sustained or paroxysmal, and patients who are diagnosed in the presymptomatic stage may have normal blood pressure. These tumors can be lethal unless they are diagnosed early and treated appropriately. Numerous disorders can mimic pheochromocytoma, leading to diagnostic dilemma. Described here is a case which was misdiagnosed for 5 years as anxiety, panic attacks, cervical spondylosis, vasomotor symptoms of menopause, arrhythmia and even acute coronary syndrome. Therefore, enhanced adrenal awareness is the need of the hour, to catch this “great masquerade”.
Keywords: Pheochromocytoma, diagnostic dilemma, metanephrines
Both pheochromocytomas and paragangliomas are rare tumors of the sympathetic nervous system. Pheochromocytomas originate from the adrenal medulla and secrete epinephrine as well as norepinephrine. Paragangliomas, or extra-adrenal pheochromocytomas, tend to originate from sympathetic paraganglia and usually show metastasis. Tumoral secretion of norepinephrine causes hypertension. Excessive epinephrine causes tachyarrhythmias. These tumors may be seen in one or both adrenals or anywhere along the sympathetic nervous chain. These tumors can be lethal and cause death in at least a third of the patients before even being diagnosed.
Catastrophic hypertensive crisis and fatal cardiac arrhythmias can occur spontaneously or may be triggered by needle biopsy or manipulation of the mass, vaginal delivery, trauma, anesthesia or surgery (both unrelated to the tumor or for its removal). Exercise, bending, lifting, defecation or emotional stress can trigger paroxysms. Certain drugs can precipitate attacks, such as decongestants, amphetamines, cocaine, epinephrine, corticosteroids, fluoxetine, metoclopramide, caffeine, nicotine and ionic intravenous contrast. Described here is a case which was misdiagnosed for 5 years as anxiety, panic attacks, cervical spondylosis, etc.
CASE REPORT
A 55-year-old normotensive female, with no significant past medical history, presented with recurrent episodes of dizziness, palpitations, paresthesia and chest pain radiating to left arm since last 5 years. The patient reported that she noticed these spells mostly while sitting on the toilet seat and during long distance traveling. The spells used to last for 1 to 2 minutes. She had visited many clinicians and was treated for anxiety, panic attacks, cervical spondylosis, vasomotor symptoms of menopause, arrhythmia and even acute coronary syndrome.
Despite her treatment, the spells persisted and her pulse and blood pressure (BP) were always found normal during and after a spell, except for an isolated home BP reading of 180/90 mmHg. Generally, the patient appeared comfortable. The system examination was benign. Her complete blood count, blood sugar, kidney functions, thyroid profile, Holter and echocardiography were normal.
A 24-hour urinary fractionated metanephrines and normetanephrines of 966 µg and 4371 µg, respectively, clinched the diagnosis. 68-Gallium (68Ga)-DOTATOC-PET (position emission tomography) scan detected a mass lesion of about 5 × 6 cm (Fig. 1), with internal necrosis abutting the right adrenal gland, with no local or distant metastasis. Diagnosis of right adrenal pheochromocytoma was made. She was started on low-dose α-blocker. A β-blocker was later added before surgery. The mass was surgically removed 1 week later.
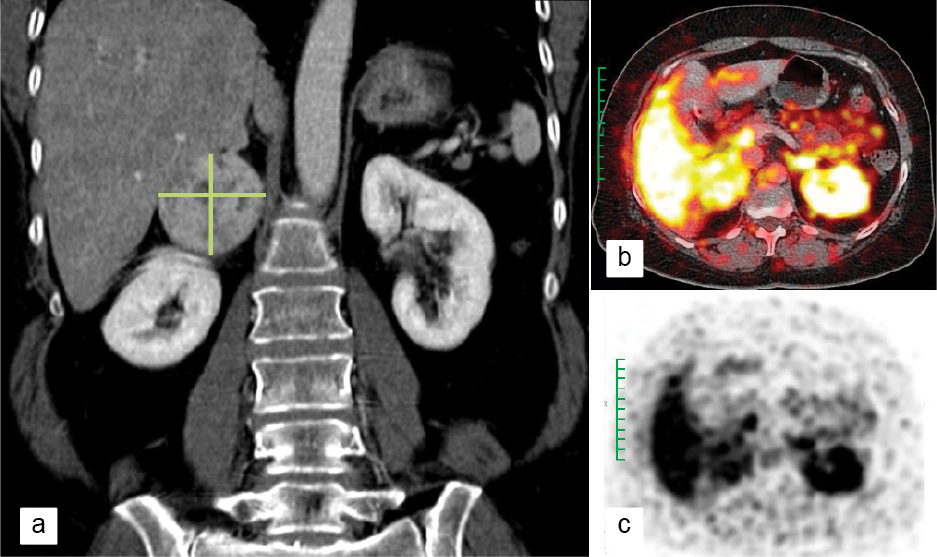
Figure 1. Right adrenal pheochromocytoma (marked in green) as seen in a) CT image, b) PET image and c) PET DOTATOC image.
DISCUSSION
Numerous disorders can cause signs and symptoms that may prompt the clinician to test for pheochromocytoma. Around 58% of the patients can have episodic nonspecific spells. Other symptoms may include anxiety, weakness/fatigue, dyspnea, tremors, sweating, dizziness, chest pain, abdominal pain or constipation. Epinephrine secretion by an adrenal pheochromocytoma can cause episodic tachyarrhythmias, orthostatic hypotension or even syncope. Due to its variable clinical presentation, pheochromocytomas have been called “the great masquerader”.
Certain conditions mimic pheochromocytoma, such as thyrotoxicosis, labile essential hypertension, myocarditis, acute coronary syndrome, arrhythmia, eclampsia, acute intermittent porphyria, hypogonadal vascular instability (hot flushes), anxiety attacks, cocaine or amphetamine use, and clonidine withdrawal. Patients taking nonselective monoamine oxidase (MAO) inhibitor antidepressants can have hypertensive crisis after eating foods that contain tyramine. Renal artery stenosis can cause severe hypertension and may coexist with pheochromocytoma.
Urinary fractionated metanephrines effectively confirm most pheochromocytomas that were detected by elevated plasma fractionated free metanephrines. Urinary assay for total metanephrines is about 97% sensitive for detecting functioning pheochromocytomas. Plasma fractionated free metanephrines can be elevated in sleep apnea or with stressful illness.
No single imaging modality is 100% sensitive for pheochromocytoma or paraganglioma. PET imaging gives crisper imaging than scintigraphy. 68Ga-DOTATOC-PET scanning is the most sensitive scan, detecting about 90% of pheochromocytomas, paragangliomas and metastases. At the time of symptom-based detection, pheochromocytomas have an average diameter of 4.5 cm.
Patients must receive adequate treatment for hypertension and tachyarrhythmias prior to surgery for pheochromocytoma/paraganglioma. Patients are advised to measure their BP daily and immediately during paroxysms. Alpha-blockers or calcium channel blockers are used, either alone or in combination. BP must be controlled prior to introducing cardioselective β-blockers for control of tachyarrhythmias.
Surgical removal of pheochromocytomas or abdominal paragangliomas is the treatment of choice. For surgery, a team approach—endocrinologist, anesthesiologist and surgeon—is critical. Laparoscopic surgery is preferred, but large and invasive tumors require open laparotomy. Prior to surgery, BP control should be maintained for a minimum of 4 to 7 days or until optimal cardiac status is established. Patients must be very closely monitored during surgery to promptly detect sudden changes in BP or cardiac arrhythmias.
CONCLUSION
Pheochromocytoma is often called “10% tumor” because 10% are bilateral, malignant, extra-adrenal, multiple, familial and occur in children. Pheochromocytoma is one of the causes of hypertension that can be surgically managed. While it is the cause of hypertension in only 0.4% of the hypertensive patients, prompt identification is essential, as it won’t just help with the management of hypertension but will also help avert the potentially life-threatening effects of the tumor.
Increasing awareness about disorders of the adrenal gland is vital. It is the first step in creating change and helps people understand the disease, encourages those with symptoms to undergo testing, and makes the emergency medical providers aware of the severity of adrenal crisis.
SUGGESTED READING
- Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SKG, Murad MH, et al. Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915-42.
- Young WF Jr, Maddox DE. Spells: in search of a cause. Mayo Clin Proc. 1995;70(8):757-65.
- Lo CY, Lam KY, Wat MS, Lam KS. Adrenal pheochromocytoma remains a frequently overlooked diagnosis. Am J Surg. 2000;179(3):212-5.
- Plouin PF, Amar L, Dekkers OM, Fassnacht M, Gimenez-Roqueplo AP, Lenders JW, et al; Guideline Working Group. European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur J Endocrinol. 2016;174(5):G1-G10.
- Soltani A, Pourian M, Davani BM. Does this patient have Pheochromocytoma? A systematic review of clinical signs and symptoms [published correction appears in J Diabetes Metab Disord. 2017;16:42]. J Diabetes Metab Disord. 2016;15:6.
- Fitzgerald PA. Pheochromocytoma & paraganglioma. In: Papadakis MA, McPhee SJ, Rabow MW (Eds.). Current Medical Diagnosis and Treatment 2021. 60th Edition, USA: McGraw Hill; 2021.