Abstract
Introduction: Rasmussen’s encephalitis (RE) is an inflammatory encephalopathy characterized by progressive refractory focal seizures, cognitive deterioration and focal neurological deficit that occur with gradual atrophy of one brain hemisphere. Case presentation: We report a case of an 18-year-old male with a history of abnormal body movements involving the right half of the body without loss of consciousness for the last 15 years. Noncontrast computed tomography (NCCT) head and magnetic resonance imaging (MRI) revealed hemiatrophy of the left cerebral hemisphere. Conclusion: RE is a rare disease; hence, diagnosing and managing such patients may be challenging. Our aim is to draw attention of the treating physicians towards this disease with the help of this case report.
Keywords: Inflammatory encephalopathy, refractory, focal, rare, abnormal body movements, epilepsia partialis continua
Rasmussen’s encephalitis (RE) is a chronic inflammatory disease of unknown origin, usually affecting one-half of brain, first described by Rasmussen et al in 1958.1 It is characterized by seizures, hemiparesis and hemiatrophy of brain. Seizures are usually focal and half of them are epilepsia partialis continua. Progressive hemiparesis and other deficits occur depending on the affected hemisphere and are usually associated with intellectual decline. Imaging of the brain shows progressive hemiatrophy.2 It affects the age group of 3 to 15 years with mean age of 6 years, and has female preponderance.3
The estimated incidence is 2.4 per 10 million people aged 18 years or younger per year.4
The etiology is unknown although autoimmune mechanism in the form of antibodies against GluR3 subunit of glutamate receptors have been identified.5
In this article, we present a case of RE as it is a rare disorder and therefore it is important to draw attention towards such cases.
CASE REPORT
An 18-year-old male patient was admitted to our medical ward with history of abnormal body movements of right half of body without loss of consciousness for last 15 years. These episodes were 1 to 2 per month and were not associated with tongue bite and urinary incontinence. The patient was not on any antiepileptic drugs. The last episode occurred 1 day prior to the present hospitalization, which was associated with loss of consciousness. Patient had episode of high-grade fever at the age of 3 years for which he was admitted at the District Hospital, where his mother noticed decreased movement of right half of body but it was not investigated. The weakness gradually worsened over the next 15 years to the extent that at present patient is unable to perform any activity like dressing, undressing, eating, brushing teeth, etc. with right upper limb resulting in muscle atrophy and contractures. Patient was born with normal vaginal delivery with no history of any complications following it. Patient had history of delayed development of milestones and poor scholastic performance.
On examination, patient was conscious and oriented to time, place and person. There was extension at right elbow joint; pronated forearm and flexion at wrist joint (Fig. 1). Mental status examination revealed impaired calculation and Mini-Mental State Examination (MMSE) revealed a score of 24/30. On cranial nerve examination, patient had right upper motor neuron (UMN) type of facial nerve palsy. On motor examination, patient had gross wasting in right upper and lower limbs. Tone was markedly increased on right side of body with reduced power (3/5), brisk deep tendon reflexes and reduced sensation to touch, pain, temperature, vibration and proprioception. Plantar response was extensor on right side. Biochemistry was unremarkable. A provisional diagnosis of insidious onset gradually progressive sensorimotor hemiparesis with focal to generalized epilepsy with mental retardation was made. Noncontrast computed tomography (NCCT) head and magnetic resonance imaging (MRI) brain was performed, which revealed left cerebral hemiatrophy as evidenced by reduction in volume, prominence of cerebral sulci and dilation of left lateral ventricle. No other bony vault abnormality or prominence of frontal and mastoid air cells were seen (Figs. 2-4). Electroencephalogram (EEG) revealed epileptiform discharges in the form of sharp and slow wave complexes. Histopathology could not be done as attendants were not willing. Patient fulfilled the diagnostic criteria of RE (Table 1). He was treated with tablet phenytoin 100 mg TDS; tablet azathioprine 50 mg OD was started to halt progression of the disease. Patient had no episode of seizure thereafter. He was advised follow-up and repeat MRI brain was planned to check for any progression.

Figure 1. Right-sided wasting with flexion deformity at wrist joint.
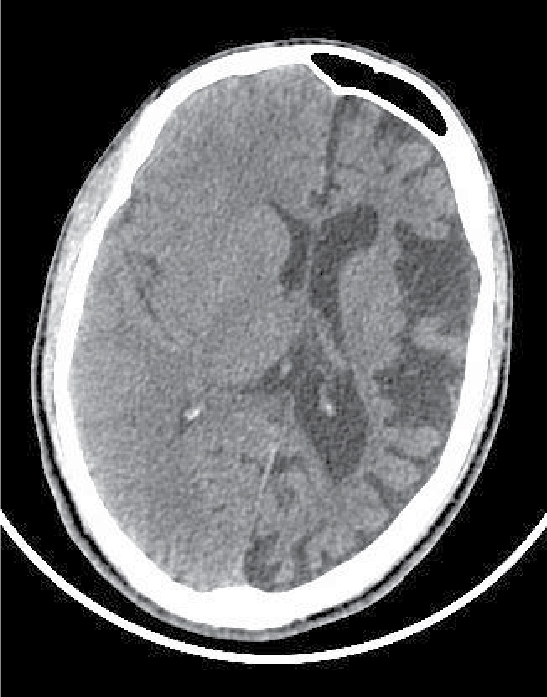
Figure 2. NCCT head showing left cerebral hemiatrophy as evidenced by reduction in volume, prominence of cerebral sulci and dilation of left lateral ventricle.
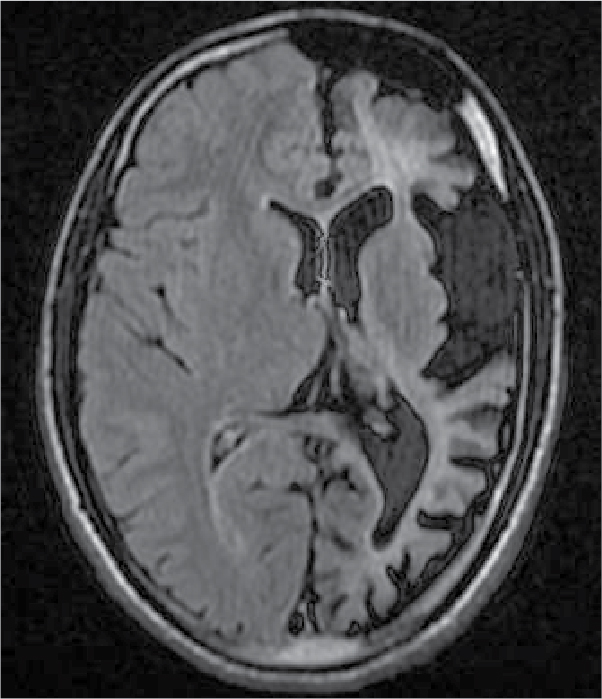
Figure 3. FLAIR transverse section showing loss of volume of white matter in left cerebral hemisphere with thinning of gyri and ex vacuo dilatation of left lateral ventricle.
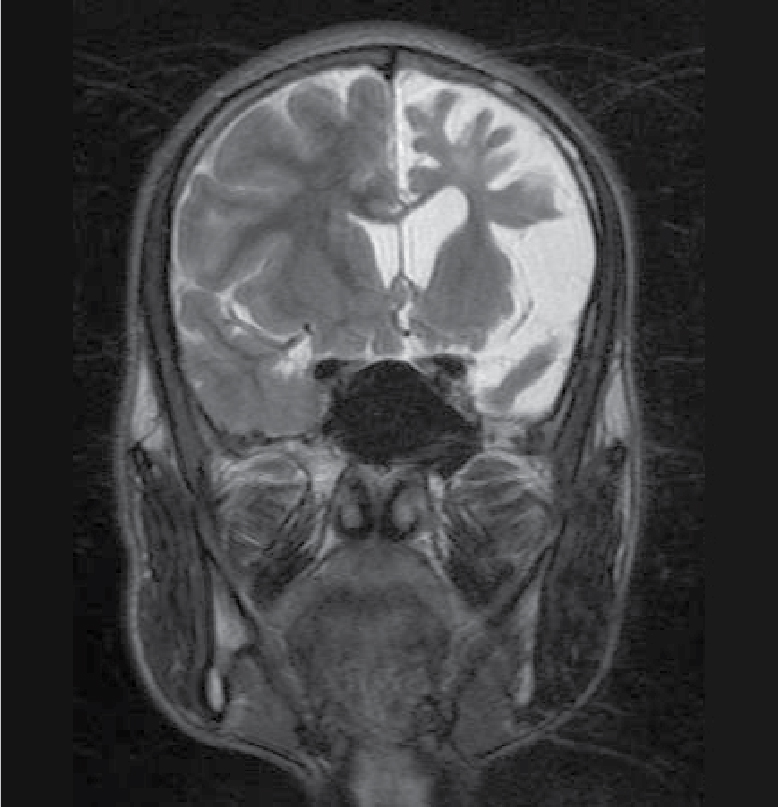
Figure 4. T2WI showing loss of volume of white matter in left cerebral hemisphere with thinning of gyri.
It is essential to differentiate RE from other causes of hemiatrophy including Dyke-Davidoff-Masson syndrome, Sturge-Weber syndrome and unihemispheric cerebral vasculitis were ruled out, both through clinical and radiological features.
DISCUSSION
Rasmussen’s encephalitis is a chronic inflammatory disease of central nervous system usually affecting one cerebral hemisphere. It is characterized by seizure, hemiparesis, hemiatrophy of brain and cognitive dysfunction. It affects children at ages 3 to 15 years, with mean age of 6 years, and occurs more often in females than in males.
Our patient presented at 18 years of age, but he had developed clinical features at the age of 5 years, which is consistent with age group within which this disease usually manifests. He also presented with similar clinical features, i.e., seizures, right-sided hemiparesis and cognitive dysfunction. The MMSE score was 24 indicating cognitive dysfunction. Brain imaging revealed left-sided hemiatrophy.
The seizures in RE remain simple partial or evolve to complex partial or secondary generalized tonic-clonic seizures, with half of them having epilepsia partialis continua. Our patient presented with simple partial seizure with epilepsia partialis continua initially and until 1 day back, when he developed secondary generalized tonic-clonic seizure.
The seizures are often intractable despite aggressive medical management.6 This is not consistent with our patient as he responded well to phenytoin.
RE has an unknown etiology, although autoimmune mechanism in the form of antibodies against GluR3 subunit of glutamate receptors has been identified.5
The diagnosis of RE is based on clinical, radiological and pathological features with emphasis on clinical and radiological features (Table 1).7
|
Table 1. Diagnostic Criteria of Rasmussen’s Encephalitis
|
|
Part A
|
|
Clinical
|
Focal seizures (may progress to epilepsia partialis continua)
Unilateral cortical defects
|
|
EEG
|
Unihemispheric slowing with or without epileptiform activity
Unilateral seizure onset
|
|
MRI
|
a: Unihemispheric focal cortical atrophy
b: Grey or white matter T2/FLAIR hyperintense signal
c: Hyperintense signal or atrophy of ipsilateral caudate head
(should include point ‘a’ and at least one among point ‘b/c’)
|
|
Part B
|
|
Clinical
|
Epilepsia partialis continua
Progressive unilateral cortical deficits
|
|
MRI
|
Progressive unihemispheric focal cortical atrophy
|
|
Histopathology
|
T-cell-dominated encephalitis with activated microglial cells and reactive astrogliosis
|
Rasmussen’s encephalitis can be diagnosed if either all three criteria of Part A or 2 out of 3 criteria of Part B are present.
Management of RE is challenging and disappointing. Steroids, immunomodulators, plasma exchange and immunoglobulins have been used in the acute phase of the disease with limited success. Surgical treatment in the form of functional hemispherectomy is needed in resistant cases. But our patient who was started on oral antiepileptics, responded well to treatment and seizures were controlled.
This case presented with classic clinical and radiological features suggestive of RE and fulfilled the diagnostic criteria as at least two criteria from Part B were met.
CONCLUSION
Rasmussen’s encephalitis is a rare disease, so diagnosing and managing such patients may pose difficulty to treating physician. With the help of this case report, our aim is to draw attention of treating physicians towards this disease. Since, it is a progressive disease; further studies need to be done for medical therapy in halting the disease process.
REFERENCES
- Rasmussen T, Olszewski J, Lloydsmith D. Focal seizures due to chronic localized encephalitis. Neurology. 1958;8(6):435-45.
- Abou- Khalil BW, Gallagher MJ, Macdonald RL. Epilepsies. In: Daroff RB, Jankovic J, Mazziotta JC, Pomeroy SL (Eds.). Bradley’s Neurology in Clinical Practice. 7th Edition. China: Elsevier Inc; 2016. pp. 1563-614.
- Ropper AH, Samules MA, Klein JP, Prasad S. Adams and Victor’s Principle of Neurology. 11th Edition. McGraw-Hill Education; 2019. pp. 332-73.
- Bien CG, Tiemeier H, Sassen R, Kuczaty S, Urbach H, von Lehe M, et al. Rasmussen encephalitis: incidence and course under randomized therapy with tacrolimus or intravenous immunoglobulins. Epilepsia. 2013;54(3):543-50.
- Rogers SW, Andrews PI, Gahring LC, Whisenand T, Cauley K, Crain B, et al. Autoantibodies to glutamate receptor GluR3 in Rasmussen’s encephalitis. Science. 1994;265(5172):648-51.
- Tien RD, Ashdown BC, Lewis DV Jr, Atkins MR, Burger PC. Rasmussen’s encephalitis: neuroimaging findings in four patients. AJR Am J Roentgenol. 1992;158(6):1329-32.
- Varadkar S, Bien CG, Kruse CA, Jensen FE, Bauer J, Pardo CA, et al. Rasmussen’s encephalitis: clinical features, pathobiology, and treatment advances. Lancet Neurol. 2014;13(2):195-205.