https://doi.org/10.59793/ijcp.v34i5.676
Abstract
Introduction: Cushing syndrome is a rare endocrine disorder caused by a variety of underlying etiologies. It can be due to exogenous or endogenous high cortisone levels (ACTH-dependent or ACTH-independent). We herein report a case of ACTH-dependent Cushing syndrome caused by pituitary and adrenal hybrid adenoma. Case report: A 42-year-old female presented with a complaint of hematemesis. She had hirsutism, central obesity and violaceous striae on her abdomen and thigh. On detailed clinical examination and relevant investigation, we found that the cause of hematemesis was esophagitis with necrotic gastric ulcer due to Cushing syndrome caused by the pituitary and adrenal hybrid adenoma. Discussion: Cushing syndrome is a rare endocrine disorder characterized by increased exogenous or endogenous serum cortisol levels, which lead to various clinical presentations. Early identification of the disease and its cause is critical. The entire clinical presentation must be considered for correct diagnosis, which is generally delayed due to the overlapping symptoms of the disease with various specialities. Conclusion: Diagnosis and management of Cushing’s syndrome continues to present considerable challenges and necessitates referral to higher centers. Its diverse presentation warrants a complete clinical, physical, radiological and endocrine examination.
Keywords: Cushing syndrome, rare, serum cortisol, ACTH-dependent, ACTH-independent, hematemesis
Cushing syndrome is a rare endocrine disease caused by excess serum cortisol, which may be due to exogenous causes such as consumption of prednisolone or endogenous causes (adrenocorticotropic hormone [ACTH]-dependent or ACTH-independent). due to a tumor of the pituitary or adrenal gland that secretes cortisol.1,2 Cushing syndrome was first described by American neurosurgeon Harvey Cushing in 1932. Around 2 to 3 people per million are affected each year.3,4 The most commonly affected age group is 20 to 50 years; females are affected 3 to 4 times more than males.
Cushing syndrome may present with varied symptomatology, which includes rapid weight gain of trunk and face with sparing of limb (central obesity), buffalo hump, moon facies, excessive sweating, thin skin, purplish reddish striae, muscle weakness, excessive hair growth (facial male pattern hair growth), baldness, amenorrhea, oligomenorrhea and infertility. Cognitive dysfunction like decreased attention, memory and depression may associated with elevated cortisol levels. Hypertension, hypokalemia, hypernatremia, infections and osteoporosis may be other important clinical presentations due to excess cortisol.5
Cushing syndrome caused by the internal production of cortisol may be of pituitary origin, which produces excessive ACTH or adrenal origin, which leads to excessive cortisol production. Around 80% to 85% of endogenous Cushing syndrome is of pituitary origin, whereas the remaining is due to hyperplastic adrenal gland or nodular adrenal hyperplasia.6
In very few cases, tumors outside the normal pituitary-adrenal axis can produce ACTH due to ectopic or paraneoplastic syndrome such as small cell carcinoma. Cushing syndrome resulting from a hybrid tumor (tumor of both pituitary and adrenal gland) is an extremely rare condition and only a handful of cases have been reported in the literature. Hence, we are reporting a case of hybrid tumor causing Cushing syndrome, admitted in our institute.1,7
CASE REPORT
A 42-year-old female housekeeper was admitted to Maharana Bhupal Government Hospital, RNT Medical College, Udaipur, Rajasthan with complaints of pain in the upper abdomen and hematemesis for the last 2 to 3 days. The frequency of hematemesis was 2 to 3 episodes/day and around 30 to 50 mL of blood in each episode. At the time of admission, the patient had tachycardia (pulse rate 130/min), hypotension (blood pressure 80/60 mmHg); the respiratory rate was 20/min. The patient was conscious with Glasgow Coma Scale score E4V5M6. The patient had anemia with hirsutism (male pattern facial hair), no cyanosis, no clubbing, no icterus and no koilonychia.
On facial examination, she had excessive facial hair (male pattern hair) (Fig. 1). The patient was clinically diagnosed with a case of suspected Cushing syndrome with hematemesis. Further workup is in the form of a detailed history, clinical examination and investigations was done. Her history revealed that this patient was hypertensive for 8 years and was on treatment with control. She underwent hysterectomy 8 years back due to irregular periods. She gave no history of taking any drug, which included corticosteroid therapy in past.
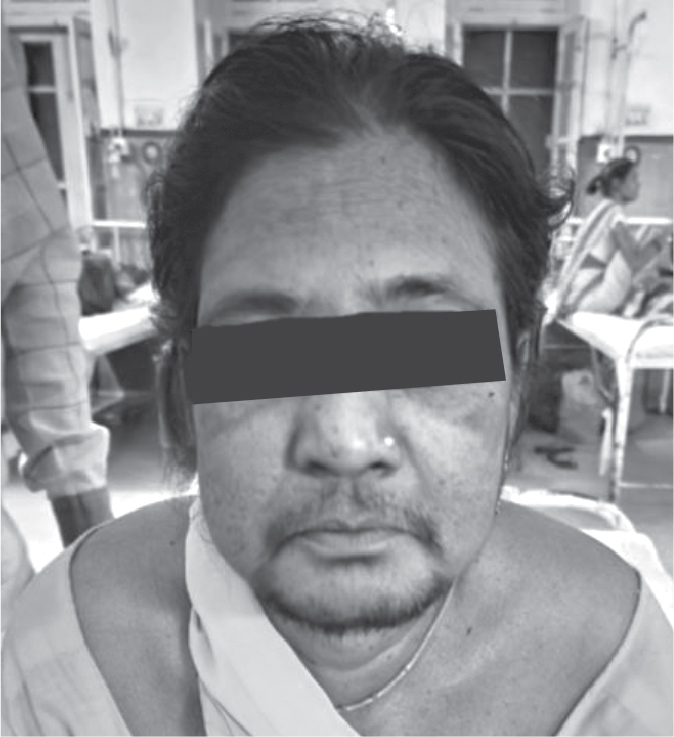
Figure 1. Showing male pattern hair on the face (hirsutism).
On detailed clinical examination, we found thin brittle hair, moon face, violet striae over the abdomen and thigh, central obesity and thin leg and arm, confirming the clinical diagnosis as Cushing syndrome.
Routine investigation showed hemoglobin - 8 g/dL, total leukocyte count (TLC) - 11,000/cu mm, platelet count - 187 lakh/cu mm, urea - 56 mg/dL, creatinine - 0.77 mg/dL, total bilirubin - 1.093 mg/dL, conjugated bilirubin - 0.33 mg/dL, serum-glutamic oxaloacetic transaminase (SGOT)/serum glutamate pyruvate transaminase (SGPT) - 19/74 U/L, triglycerides - 175 mg/dL, high-density lipoprotein (HDL) cholesterol - 29 mg/dL, low-density lipoprotein (LDL) cholesterol - 60 mg/dL. Serum electrolytes were sodium - 148 mEq/L, potassium - 3.8 mEq/L and chloride - 117 mEq/L. Her viral markers (HBV, HCV, HIV) were also negative.
Her hormone assays showed triiodothyronine (T3) - 0.5 nmol/L (1.3-3.1), thyroxine (T4) - 43.6 nmol/L (66-181), thyroid-stimulating hormone (TSH) - 0.3 uIU/mL (0.27-4.20), follicle-stimulating hormone (FSH) - 1.6 mIU/mL (1-6), luteinizing hormone (LH) - 0.3 mIU/mL (3-13), prolactin - 262 uIU/mL (155-324), testosterone - 1.56 nmol/l (0.5-2.4), serum cortisol (early morning) - 874.8 nmol/L (138-635). The dexamethasone overnight suppression test was found positive (not suppressed)6 serum ACTH was 48.6 pg/mL.
Her USG abdomen was showing an absent uterus and nonvisualization of both ovaries with normal suprarenal glands.
Upper gastrointestinal endoscopy revealed esophagitis with necrosed gastric mucosa (Fig. 2).
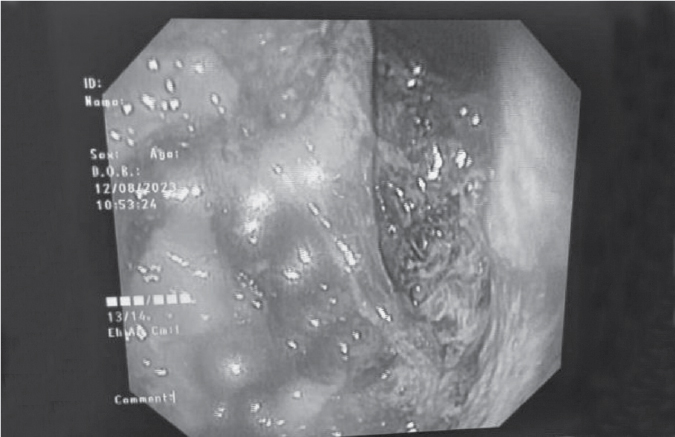
Figure 2. Upper gastrointestinal endoscopy of the patient showing esophagitis with necrosed gastric mucosa.
Computed tomography (CT) scan of the abdomen showed a soft tissue density lesion measuring 15 ´ 14 mm (Hounsfield Unit [HU] ~4-5) in the medial limb of the right adrenal gland (Fig. 3).
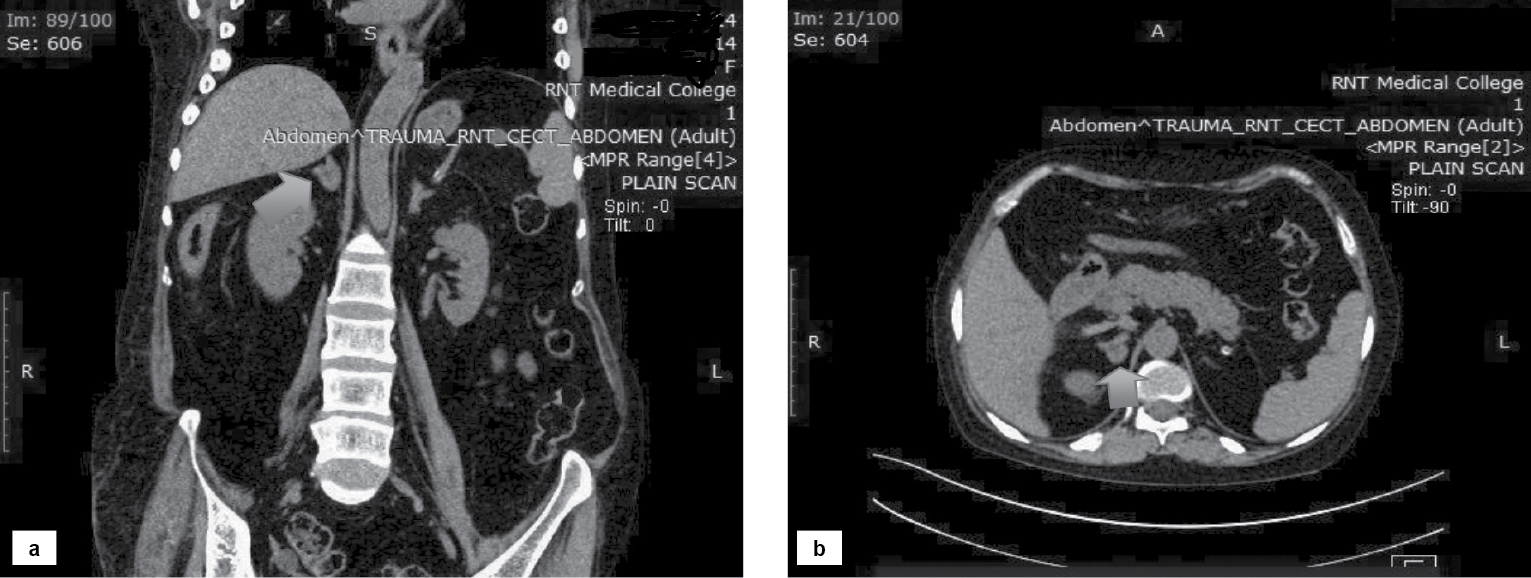
Figure 3. Coronal (a) and transverse (b) sections showing soft tissue density lesion in the medial limb of the right adrenal gland.
Magnetic resonance imaging (MRI) scan of the pituitary gland with contrast showed a hypointense lesion of size measuring approximately 6 ´ 5 mm is seen involving the right side of the pituitary gland. There is no extension above sella turcica suggestive of pituitary microadenoma (Fig. 4).
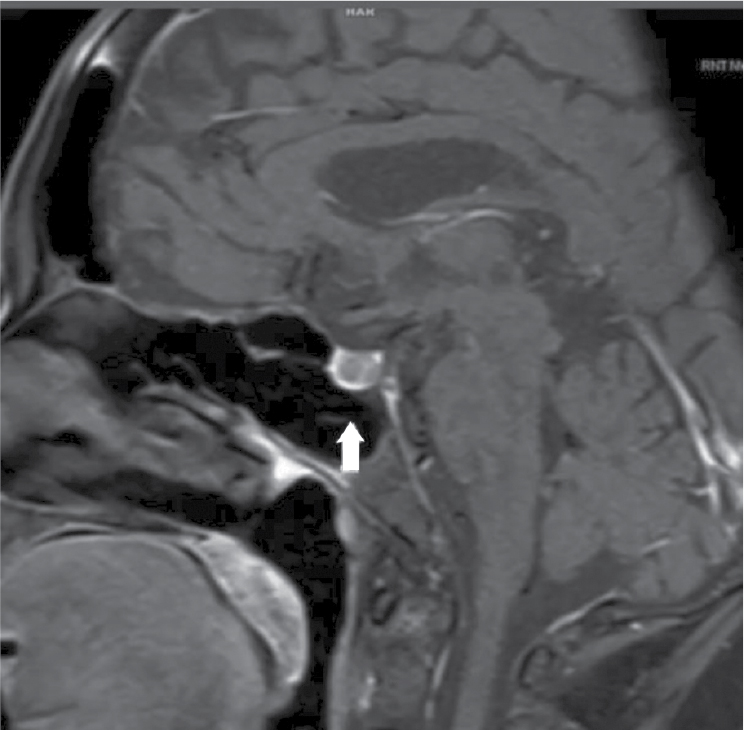
Figure 4. MRI pituitary with contrast showing hypointense lesion involving the right side of the pituitary gland. There is no significant enhancement in post-contrast. There is no extension above sella turcica suggestive of microadenoma.
After all the appropriate investigations and imaging, the cause of Cushing syndrome was found to be a pituitary and adrenal hybrid tumor. The patient was treated symptomatically; blood transfusions and a proton pump inhibitor (PPI) with electrolyte correction were given. Neurosurgery and urology opinions were taken and advised surgical intervention but unfortunately, the patient had bouts of massive hematemesis and she died.
DISCUSSION
Cushing syndrome is a rare disease characterized by increased levels of glucocorticoids manifesting as the upregulation of gluconeogenesis, lipolysis and protein catabolism.
The causes of Cushing’s syndrome can be divided into two categories. The first category includes ACTH-dependent Cushing’s syndrome, which includes Cushing’s disease (ACTH-producing pituitary adenoma, ectopic ACTH syndrome).2 Ectopic ACTH syndrome is due to ACTH secretion by bronchial and pancreatic carcinoid tumors, small cell lung carcinoma, medullary carcinoma thyroid, pheochromocytoma, etc.8 The second category includes ACTH-independent Cushing’s syndrome, which includes adrenocortical adenoma, adrenocortical carcinoma and micro- and macronodular adrenal hyperplasia. Exogenous Cushing’s syndrome is more common than the endogenous Cushing’s syndrome. Sometimes mild cortisol excess due to the iatrogenic prescription of steroids as drug treatment for autoimmune or autoinflammatory conditions leads to Cushing syndrome.
The diagnosis of Cushing syndrome is a difficult task. The initial screening tests include a 24-hour-urinary-free cortisol level in three separate collections, dexamethasone overnight suppression test and midnight salivary cortisol level.9 If any 1 out of the 3 is found to be positive then we go for differential diagnosis testing. Plasma ACTH levels in ACTH-dependent Cushing’s syndrome are >15 pg/mL, while they are <5 pg/mL in ACTH-independent Cushing’s syndrome.9 Depending upon the plasma ACTH level we go ahead with radiological imaging.
We found that the dexamethasone overnight suppression test was positive in our patient. The plasma ACTH level is found to be >15 pg/mL (48.6 pg/mL). In our case, we proceeded with imaging, i.e., MRI pituitary with contrast and noncontrast CT adrenal protocol. Surprisingly, we found a pituitary microadenoma with adrenal adenoma, which leads to the rarity of our case.
The coexistence of adrenal adenoma and pituitary adenoma with Cushing’s syndrome is a rare possibility. Due to its diverse presentation, an accurate clinical physical and endocrine examination is always recommended. Similar to current cases, early diagnosis may be delayed due to a variety of clinical presentations and the referral to different specialities based on their dominant symptoms.
CONCLUSION
The coexistence of adrenal adenoma and pituitary adenoma with Cushing’s syndrome is a rare possibility. Due to the diversity in the presentation of Cushing’s syndrome an accurate clinical, physical and endocrine examination is always recommended.
Financial Disclosure: No financial aid was taken from any party.
REFERENCES
- Ahmed SF, Bapir R, Fattah FH, Mahmood AG, Salih RQ, Salih AM, et al. Simultaneous pituitary and adrenal adenomas in a patient with non ACTH dependent Cushing syndrome; a case report with literature review. Int J Surg Case Rep. 2022;94:107038.
- Duan K, Gomez-Hernandez K, Mete O. Republished: Clinicopathological correlates of adrenal Cushing’s syndrome. Postgrad Med J. 2015;91(1076):331-42.
- Ellis H. Harvey Cushing: Cushing’s disease. J Perioper Pract. 2012;22(9):298-9.
- Steffensen C, Bak AM, Rubeck KZ, Jørgensen JO. Epidemiology of Cushing’s syndrome. Neuroendocrinology. 2010;92(Suppl 1):1-5.
- Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart PM, et al. The diagnosis of Cushing’s syndrome: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2008;93(5):1526-40.
- Newell-Price J, Bertagna X, Grossman AB, Nieman LK. Cushing’s syndrome. Lancet. 2006;367(9522):1605-17.
- Hiroi N, Chrousos GP, Kohn B, Lafferty A, Abu-Asab M, Bonat S, et al. Adrenocortical-pituitary hybrid tumor causing Cushing’s syndrome. J Clin Endocrinol Metab. 2001;86(6):2631-7.
- Beuschlein F, Hammer GD. Ectopic pro-opiomelanocortin syndrome. Endocrinol Metab Clin North Am. 2002;31(1):191-234.
- Wood PJ, Barth JH, Freedman DB, Perry L, Sheridan B. Evidence for the low dose dexamethasone suppression test to screen for Cushing’s syndrome—recommendations for a protocol for biochemistry laboratories. Ann Clin Biochem. 1997;34(Pt 3):222-9.