Abstract
Introduction: Dyke-Davidoff-Masson syndrome (DDMS) is characterized by seizures, facial asymmetry, contralateral hemiplegia and mental retardation. The characteristic radiological features are cerebral hemiatrophy with homolateral hypertrophy of skull and sinuses. Case report: We report a case of DDMS in a 41-year-old female who presented with generalized tonic-clonic seizures, hemiparesis of the right upper and lower limb with deviation of the mouth to left. Noncontrast computed tomography (NCCT) head and magnetic resonance imaging (MRI) revealed hemiatrophy of the right cerebral hemisphere. Conclusion: DDMS is a rare disease; hence, diagnosing and managing such patients may be challenging. Our aim is to draw attention of the treating physicians towards this disease with the help of this case report.
Keywords: Tonic-clonic seizures, hemiparesis, facial asymmetry, cerebral hemiatrophy, homolateral hypertrophy of skull, NCCT head
Dyke-Davidoff-Masson syndrome (DDMS) was initially identified in 1933 by Dyke, Davidoff
and Masson. They reported the pneumoencephalographic and plain skull radiography abnormalities in a group of 9 individuals.1 Nearly 100 cases have been reported since it was first characterized in 1933,2 although only 21 cases involving adults have been documented.3 Recurrent seizures, facial asymmetry, contralateral hemiplegia, mental retardation or learning difficulty, and speech and language problems are the most prevalent presentations. Sensory loss and mental health issues like schizophrenia are seldom reported.4,5 Cerebral hemiatrophy with ipsilateral compensatory enlargement of the skull and sinuses are characteristic radiological findings. Most cases of the syndrome in adults and adolescents have been reported.6,7
In this article, we present a case of DDMS as it is a rare disorder and therefore it is important to draw attention towards such cases.
CASE REPORT
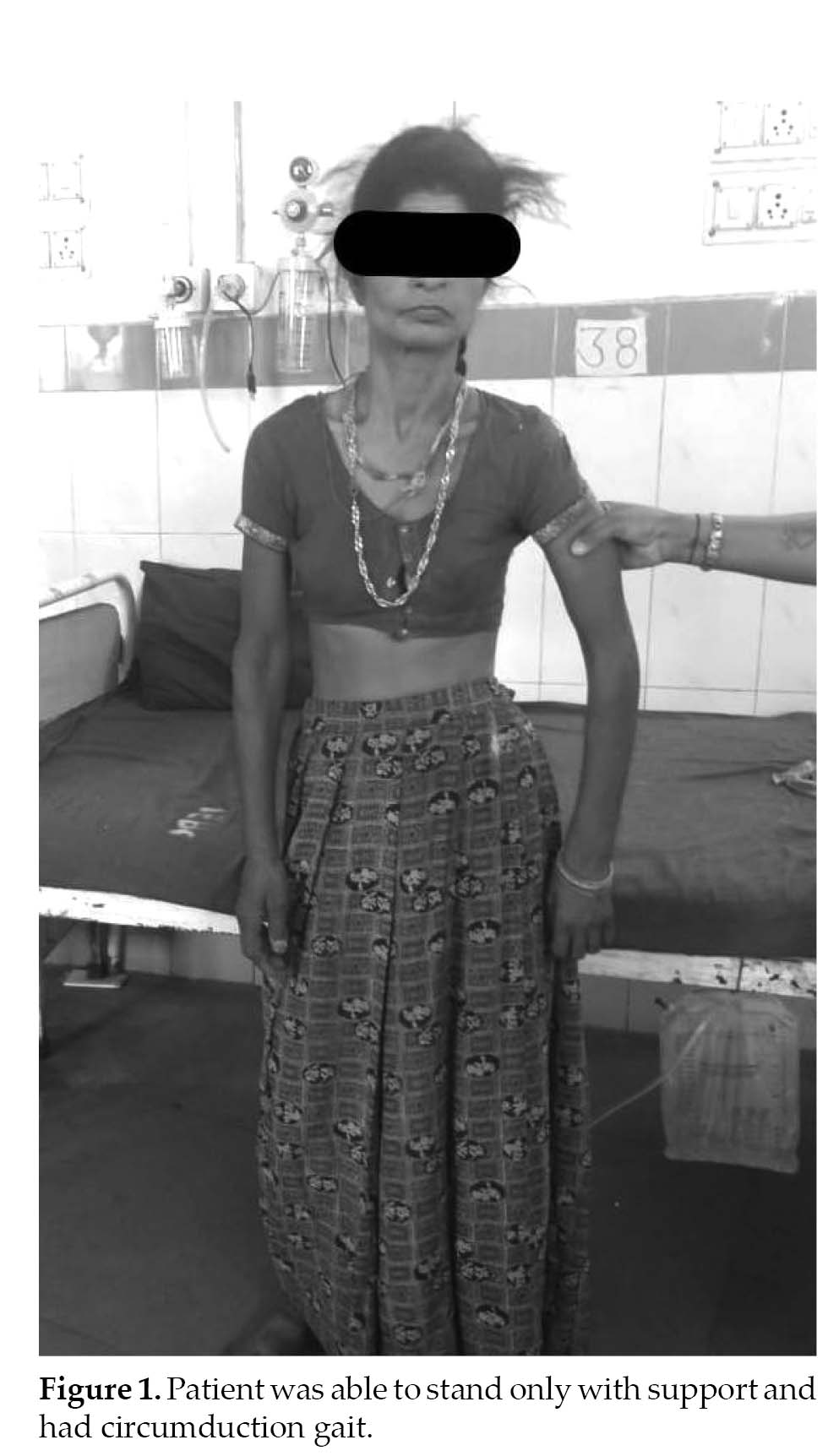
A 41-year-old right-handed woman, presented in emergency ward with 3 to 4 episodes of sudden-onset of abnormal movements (tonic-clonic) since 2 days involving all 4 limbs associated with deviation of angle of mouth to left side and frothing from mouth and urinary incontinence. This was followed by altered sensorium, which was nonprogressive, slurring of speech and weakness in right upper and lower limb, which was maximum at the time of onset and nonprogressive (Fig. 1).
Patient had a past history of similar complaint of abnormal body movement and poor scholastic performance since childhood. She was receiving tablet phenytoin 100 mg thrice a day but there was no computed tomography (CT)/magnetic resonance imaging (MRI)/electro-encephalogram (EEG) report available. Patient was born with normal vaginal delivery with no history of complication following it. Patient had history of delayed developmental of milestones and poor scholastic performance.
On examination, patient was conscious and not oriented to time, place and person initially because of postictal confusion but later she regained her orientation within a day. Mental status examination revealed a score of 22/30. Cranial nerve examination revealed right upper motor neuron type of facial palsy. Patient’s muscle tone was markedly reduced and power was 3/5 in right upper and lower limb, while left upper and lower limb
motor examination was normal. Deep tendon reflexes were brisk and plantar response was extensor. Sensory examination was normal.
Lab biochemistry was unremarkable. A provisional diagnosis of sudden-onset generalized epilepsy with motor hemiparesis and mental retardation was made.
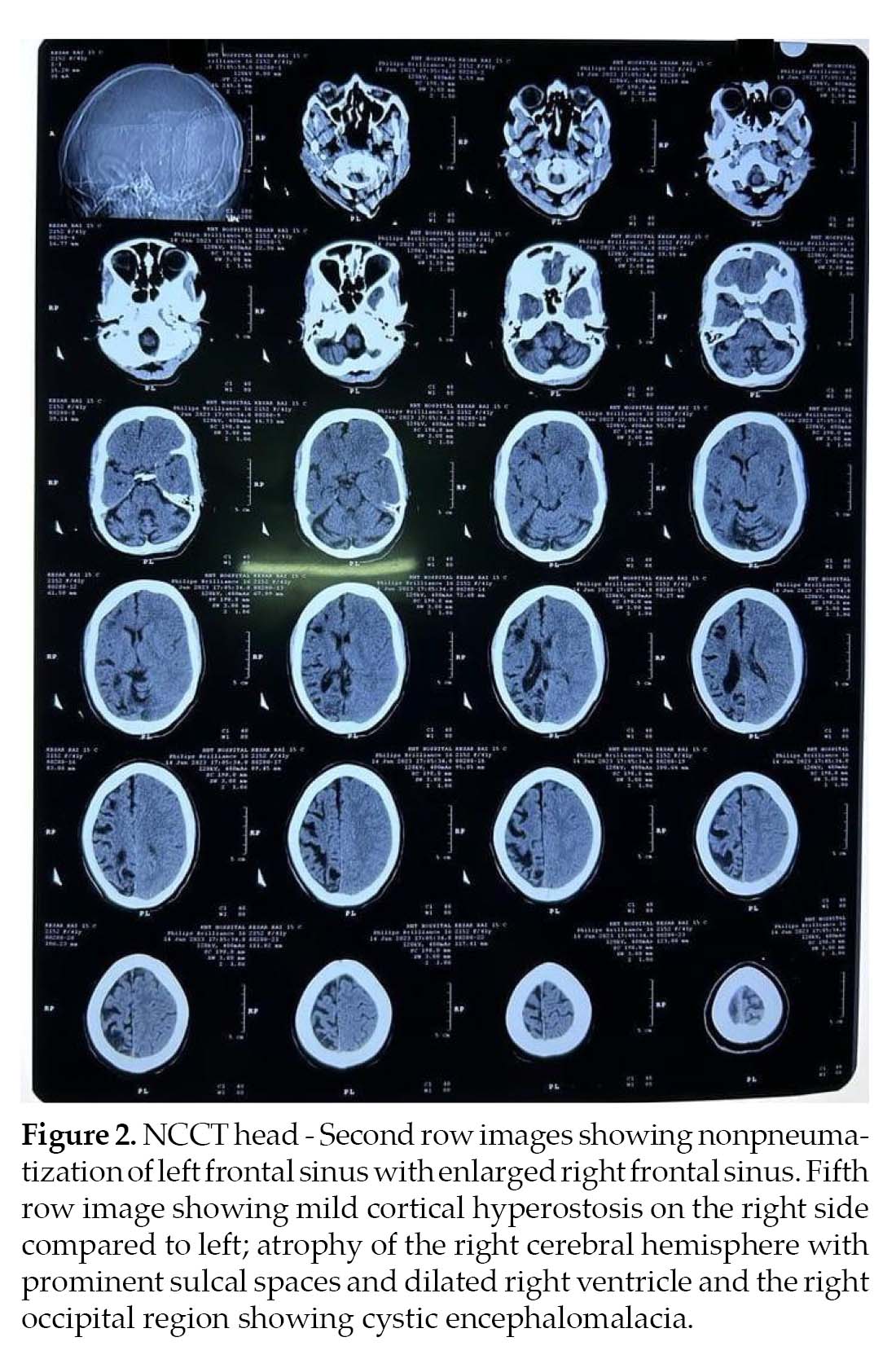
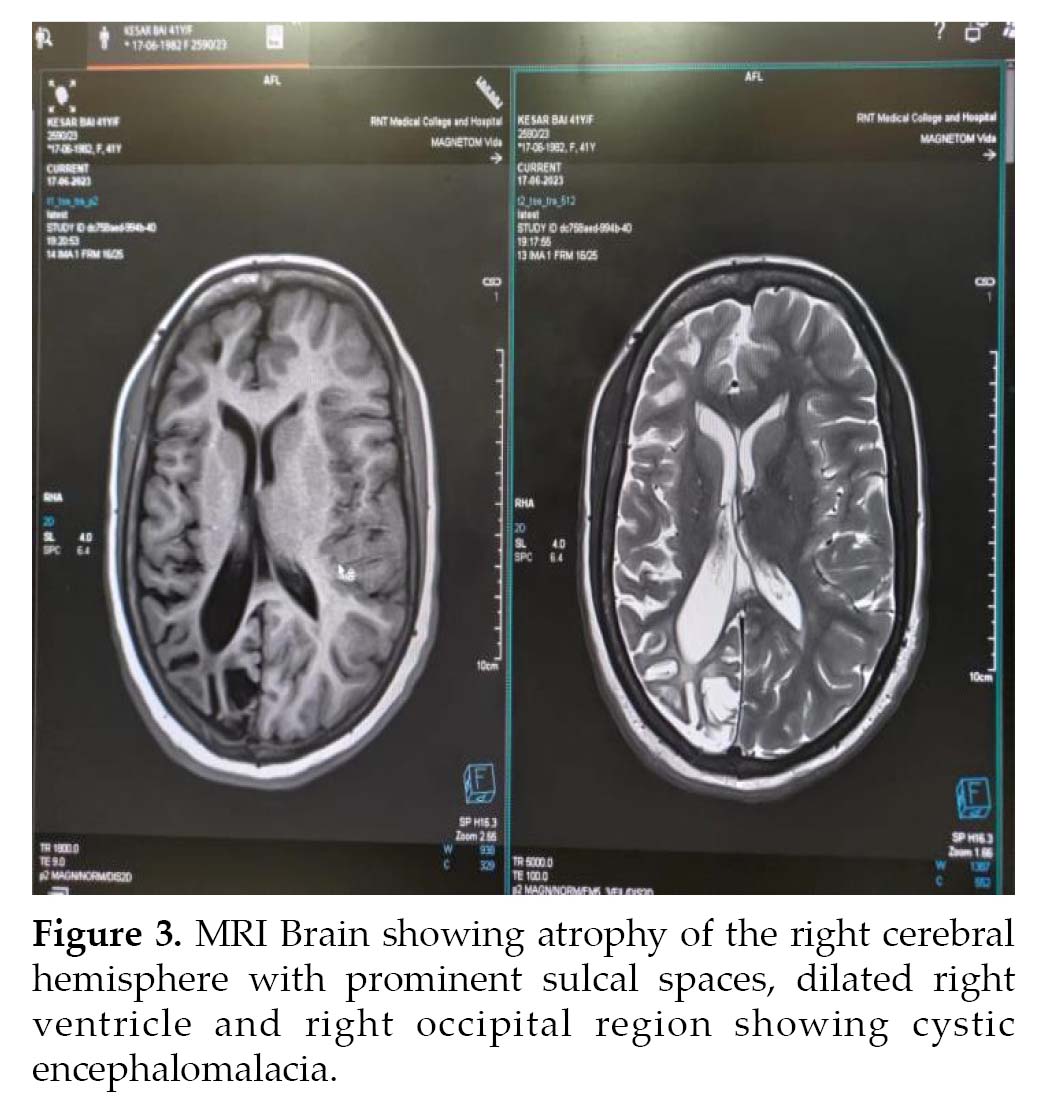
Noncontrast computed tomography (NCCT) (Fig. 2) head and MRI (Fig. 3) brain was performed, which revealed atrophy and prominent sulci and gyri of right cerebrum and cerebellum, with dilation of right lateral ventricle. There was mild cortical hyperostosis on the right side compared to the left. Nonpneumatization of left frontal sinus with enlarged right frontal sinus was seen. The left occipital region showed cystic encephalomalacia. EEG revealed epileptiform discharges in the form of paroxysmal generalized spike and wave having amplitude of 100-200 µV. Histopathology could not be done as the attendants were not willing. The patient was treated with tablet phenytoin 300 mg HS and tablet valproate 500 mg BD.
DISCUSSION
The uncommon neurological disorder known as DDMS was first identified in 1933 in 9 patients presenting with hemiparesis, facial asymmetry, seizures, intellectual disability and skull radiography findings of pneumatoencephalographic changes.8 The exact cause of Down syndrome is still unknown, but prior research has linked the disorder to a brain injury that occurred either during pregnancy (congenital subtype), which manifests in infancy or in early childhood (acquired subtype), which may have resulted from a variety of causes including trauma, ischemia, hemorrhage or infection.2 While DDMS is commonly thought of as a childhood illness, adult occurrences have been documented; a recent review found only 21 cases in the adult population.2
When the injury happens in utero, it results in congenital hemiatrophy, which is characterized by a shift of midline structures to the side of the illness and the absence of the sulcal prominence that would normally replace the gliotic tissue.9 This characteristic sets it apart from early-life cerebral hemiatrophy. Trauma, inflammation or vascular malformations, and occlusions have all been proposed as the etiological factors of DDMS. Gestational vascular occlusion, which mainly affects the middle cerebral vascular territory, may be the cause of the insult when it happens in utero. Several Indian studies have reported a potential etiological relationship between seizures and cerebral hemiatrophy.10,11
The following conditions are included in the differential diagnosis of DDMS: Rasmussen encephalitis, Fishman syndrome, linear nevus syndrome, Sturge-Weber syndrome and basal cell germinoma.12 The accurate diagnosis is given by a complete clinical history and the results of a CT or MRI. Treatment for convulsions, hemiplegia, hemiparesis and learning disabilities is symptomatic. If hemiparesis develops after the age of 2 years and there are no prolonged or recurrent seizures, the prognosis is better. Hemispherectomy has an 85% success rate in carefully chosen cases and is a potential treatment option for children with hemiplegia and intractable disabilities.13
CONCLUSION
DDMS stands as a rare neurological condition first identified in 1933. While its prevalence remains low, with nearly 100 cases reported to date, it presents with distinct clinical features including recurrent seizures, facial asymmetry, contralateral hemiplegia and cognitive impairments. Despite its rarity, the syndrome warrants attention due to its potential impact on patient quality of life. By presenting a case of DDMS in this article, we contribute to the growing body of knowledge surrounding this condition, emphasizing the importance of recognizing and addressing rare disorders in clinical practice. Increased awareness and understanding of DDMS may facilitate earlier diagnosis and intervention, ultimately improving outcomes for affected individuals.
REFERENCES
- Dyke CG, Davidoff LM, Masson LB. Cerebral hemiatrophy with homolateral hypertrophy of skull and sinuses. Surg Gynecol Obstet. 1933;57:588-600.
- Wang B, Jiang W, Yan W, Tian J, Xu J, Li Y, et al. Clinical characteristics and neuroimaging findings of seven patients with Dyke Davidoff Masson syndrome. BMC Neurol. 2021;21(1):213.
- Diestro JDB, Dorotan MKC, Camacho AC, Perez-Gosiengfiao KT, Cabral-Lim LI. Clinical spectrum of Dyke-Davidoff-Masson syndrome in the adult: an atypical presentation and review of literature. BMJ Case Rep. 2018;2018:bcr2018224170.
- Ono K, Komai K, Ikeda T. Dyke-Davidoff-Masson syndrome manifested by seizure in late childhood: a case report. J Clin Neurosci. 2003;10(3):367-71.
- Amann B, García de la Iglesia C, McKenna P, Pomarol-Clotet E, Sanchez-Guerra M, Orth M. Treatment-refractory schizoaffective disorder in a patient with Dyke-Davidoff-Masson syndrome. CNS Spectr. 2009;14(1):36-9.
- Singh P, Saggar K, Ahluwalia A. Dyke-Davidoff-Masson syndrome: classical imaging findings. J Pediatr Neurosci. 2010;5(2):124-5.
- Koshy B, Surendrababu NR. Image in medicine. Dyke-Davidoff-Masson syndrome. Ann Acad Med Singap. 2010;39(6):501-2
- Abdul Rashid AM, Md Noh MSF. Dyke-Davidoff-Masson syndrome: a case report. BMC Neurol. 2018;18(1):76.
- Sener RN, Jinkis JR. MR of craniocerebral hemiatrophy. Clin Imaging. 1992;16(2): 93-7.
- Nair KP, Jayakumar PN, Taly AB, Arunodya GR, Swamy HS, Shanmugam V. CT in simple partial seizures in children: a clinical and computed tomography study. Acta Neurol Scand. 1997;95(4):197-200.
- Garg RK, Karak B. Cerebral hemiatrophy: a possible etiological relation with fertile seizures. Indian Pediatr. 1998;35:79-81.
- Rao KCVG. Degenerative diseases and hydrocephalus. In: Lee SH, Rao KDVG, Zimmerman RA (Eds.). Cranial MRI and CT. 4th Edition. New York: McGraw-Hill; 1999. pp. 212-4.
- Narain NP, Kumar R, Narain B. Dyke-Davidoff-Masson syndrome. Indian Pediatr. 2008;45:927-8.