Abstract
This case presents a rare co-occurrence of Charcot-Marie-Tooth disease type 2 (CMT2) and schwannoma, shedding light on the intricate relationship between two neurogenic disorders. CMT is a hereditary sensorimotor peripheral neuropathy while schwannoma is a benign encapsulated tumor involving Schwann cells. The co-occurrence has been previously described in 2 patients in a family with CMT type 1A. Our case reports the first documented instance of the co-occurrence of bilateral hypoglossal schwannoma in CMT2.
Keywords: Hereditary motor and sensory neuropathy, axonal neuropathy, CMT2, schwannoma, bilateral hypoglossal schwannoma
Charcot-Marie-Tooth disease (CMT) is the most common inherited neurological disorder that affects sensory and motor peripheral nerves leading to sensory loss and progressive muscle weakness. Inheritance pattern can be autosomal dominant/autosomal recessive/X-linked recessive (AD/AR/XLR) depending on the specific gene involved. These disorders cause demyelination, axonal loss, and abnormal interactions between Schwann cells and the axons they ensheath. The rare combination of CMT1A and median nerve and spinal cord schwannomas have been previously reported1. Our case is the first documented case of bilateral hypoglossal schwannoma in CMT2.
CASE HISTORY
A 41-year-old female developmentally normal till 15 years of age presented with insidious onset gradually progressive symmetric, distal more than proximal, weakness of all four limbs, lower limbs followed by upper limb involvement, with wasting (Fig. 1) and foot deformities. Sensory examination revealed graded sensory loss of all modalities (both small fiber and large fiber) with impaired joint position and vibration sense with no peripheral nerve thickening and spinal deformities with no significant family history with normal higher mental function, cranial nerves including hypoglossal (Fig. 2) (with no atrophy, weakness, or fibrillations), cerebellum, and autonomic functions. In view of the chronic long-standing slowly progressive distal prominence neuropathy with pes cavus (Fig. 3) and hammer toes (Fig. 4), the possibility of hereditary sensory and motor neuropathy was considered. On investigating complete blood count (CBC), random blood sugar (RBS), renal function test (RFT), liver function test (LFT), serum electrolytes, serum B12, thyroid profile, autoimmune panel, voluntary counseling and testing center (VCTC), split skin smear for leprosy were negative. Serum electrophoresis and urine Bence-Jones protein, paraneoplastic workup and other relevant investigations to exclude acquired pathology were negative.
Nerve conduction study revealed bilateral lower limb sensory-motor axonal and bilateral upper limb sensory axonal polyneuropathy2. As a part of routine investigation and to exclude structural pathology, magnetic resonance imaging (MRI) brain with spine contrast was done, which revealed two well-defined T1 hypointense and T2 hyperintense mildly enhancing lesions of size 7 × 4 mm on right and 8 × 4 mm in left lateral cerebellomedullary cisterns with no diffusion restriction or mass effect over adjacent structures suggestive of bilateral hypoglossal schwannoma (Fig. 5 a and b)3. Sural nerve biopsy showed severe axonal neuropathy with no granulomas, inflammation, storage material, or amyloidosis (Fig. 6). Fite-Faraco stain for Mycobacterium leprae was negative. CMT diagnosis is a comprehensive approach that involves clinical findings, electrodiagnostic findings and genetic analysis for specific gene mutations.
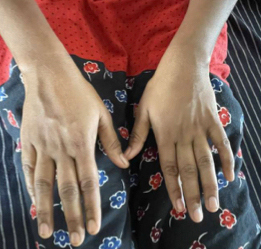
Figure 1. Illustrates small muscle wasting of hand.
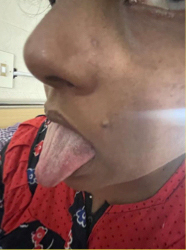
Figure 2. No atrophy of tongue.
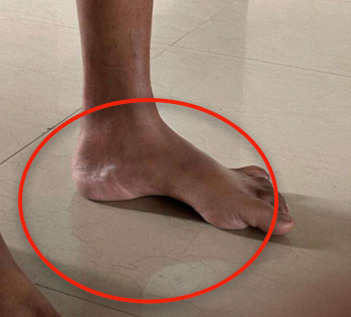
Figure 3. Illustrates pes cavus.
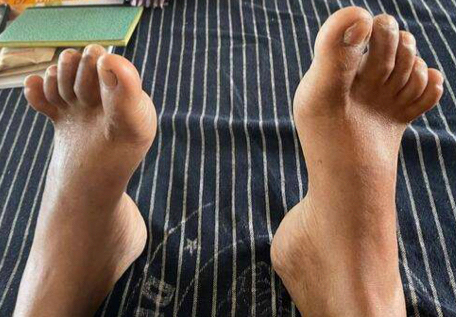
Figure 4. Illustrates hammer toes.
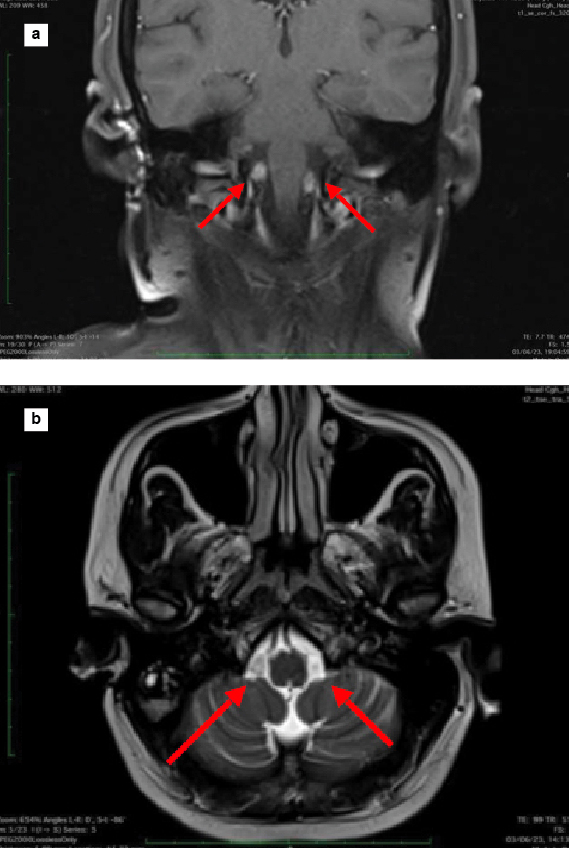
Figure 5 a and b. MRI T1 coronal contrast (a) and T2 axial (b) showing two well-defined enhancing lesions in lateral cerebellomedullary cisterns suggestive of hypoglossal schwannoma.
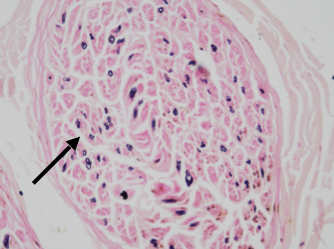
Figure 6. Histopathological examination of right sural nerve showing severe axonal loss with Kulchitsky stain.
With this clinical background of slowly progressive course that did not lead to severe disability, electrodiagnostics (upper limb motor nerve conduction velocities >45 m/s) and nerve biopsy showed predominant axonal involvement; hence, the probability of CMT2 was considered in our case4. Due to logistic reasons, genetic analysis was planned during follow-up.
DISCUSSION
Charcot-Marie-Tooth disease and schwannomas are distinct neurological conditions with limited reported associations. Heckmann et al reported a case of hereditary neuropathy with liability to pressure palsy (HNPP) in which PMP22 gene deletion is demonstrated5. A case report on schwannomas of spinal cord and median nerve have been reported in 2 patients in a family with CMT1A with PMP22 gene duplication1. These case reports suggested that the dysfunction of PMP22 gene may not only result in peripheral neuropathy but also schwannomas providing a common pathogenic basis for Schwann cell abnormality. However, to the best of our knowledge, after reviewing the available literature no case has been reported in CMT2. This is the first documented case in CMT2. Several genes involved in CMT2 includes MFN2 (Mitofusin 2), GARS (Glycyl-
tRNA synthetase), NEFL (Neurofilament light chain), HSPB1 (Heat shock protein beta 1), MPZ (Myelin protein zero), and several other genes. Further studies are needed to shed light on the exact pathophysiology behind shared pathogenesis of schwannoma and CMT2.
CONCLUSION
This is the first case report on bilateral hypoglossal schwannoma in a patient with CMT2 despite lacking genetic analysis due to the rarity of this co-occurrence.
Further genetic research is needed to study the occurrence of schwannomas in CMT2 and that would guide us to establish optimal management settings for affected patients.
REFERENCES
- Kwon JY, Chung KW, Park EK, Park SW, Choi BO. Charcot-Marie-Tooth 1A concurrent with schwannomas of the spinal cord and median nerve. J Korean Med Sci. 2009;24(4):763-6.
- Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain. 1980;103(2):259-80.
- Lee SE, Park SW, Ha SY, Nam TK. A case of cauda equina syndrome in early-onset chronic inflammatory demyelinating polyneuropathy clinically similar to charcot-marie-tooth disease type 1. J Korean Neurosurg Soc. 2014;55(6):370-4.
- Banchs I, Casasnovas C, Albertí A, De Jorge L, Povedano M, Montero J, et al. Diagnosis of Charcot-Marie-Tooth disease. J Biomed Biotechnol. 2009;2009:985415.
- Heckmann JG, Dütsch M, Buslei R. Hereditary neuropathy with liability to pressure palsy combined with schwannomas of the median and medial plantar nerves. Muscle Nerve. 2007;35(1):122-4.