Spontaneous
intracranial hypotension (SIH) is an uncommon condition characterized by low
cerebrospinal fluid (CSF) volume through a dural defect leakage, which results
in multiple debilitating clinical manifestations. Although ocular
manifestations are common, vision impairment due to SIH is not commonly
reported. Optic neuropathy is a rare yet significant complication of SIH. This
case documents the association between optic neuropathy and SIH, highlighting
the diagnostic challenges and the complexities of managing this condition.
Keywords: Orthostatic headache, bilateral optic neuropathy,
subdural hygroma, dural venous thrombosis, epidural blood patch
The clinical characteristics of spontaneous intracranial
hypotension (SIH) often result from stretching of the intracranial and cervical
dura and dilatation of intracranial venous structures and dural sinuses
resulting in orthostatic headache, neck pain, and radicular symptoms1.
The common ocular manifestations include blurry vision, double vision, and
ophthalmoparesis2. Visual field deficits are related to stretching
of optic nerve over pituitary fossa or vascular congestions of the optic nerve.
Optic neuropathy in SIH is a rare and underreported phenomenon. Previously one
case reported an association between monocular optic neuropathy and SIH1.
This is the first case documenting the association of bilateral optic
neuropathy in SIH and sheds light on this infrequent complication.
A 21-year-old female with no
known comorbidities presented with the complaints of insidious-onset, gradually
progressive orthostatic headache for 1 year. The headache was holocranial,
throbbing type of moderate to severe intensity associated with neck pain.
Headache had gradually evolved into nonorthostatic chronic, persistent headache for 5 months. The headache was triggered by coughing and abrupt head movements. She also noticed gradually progressive painless diminution of vision of both eyes
(right more than left [R > L]) in the last 4 months with no history of diplopia/lower cranial nerve involvement. There was no relevant medical history or previous trauma or lumbar puncture. On examination, patient was moderately built and nourished.
There were no cutaneous or articular stigmata for connective tissue disorder. Cranial nerve examination revealed right eye relative afferent pupil defect (RAPD) (Fig. 1) with perception of light in right eye and visual acuity of 1/60 in left eye.
Fundus examination revealed temporal pallor of optic disc (R > L) with well-defined margins with normal retinal vessels with dull macular foveal reflex suggestive of both eye primary optic neuropathy (R > L) (Fig. 2). Ocular movements were full;
examination of lower cranial nerves, sensorimotor, cerebellum, and higher mental functions were unremarkable. On investigating, complete blood picture revealed microcytic hypochromic anemia; renal function tests, liver function tests, thyroid profile,
urine routine were within limits. Voluntary counseling and testing for human immunodeficiency virus (HIV) was negative. Antinuclear antibody test, serum homocysteine, antiphospholipid antibody, serum angiotensin-converting enzyme were negative.

Figure 1. Right eye RAPD.
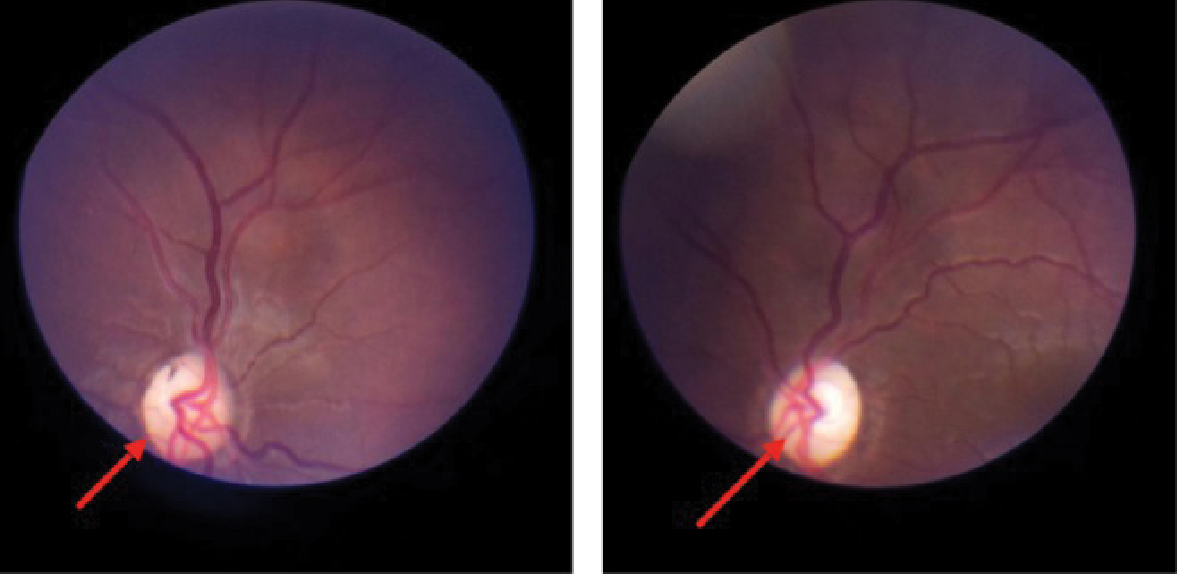
Figure 2. Both eye fundus - pale disc
with well-defined margins, normal retinal vessels suggestive of primary optic
neuropathy.
Optical coherence tomography revealed thinning of
retinal nerve fiber layer of all quadrants in right eye, and superotemporal and
inferotemporal quadrants in the left eye. A visual evoked potential (VEP)
revealed no response in the right eye and reduced amplitude and prolonged P100
latency in left eye. A lumbar puncture was performed and the cerebrospinal
fluid (CSF) opening pressure measured at the lateral decubitus position was 5 cm of H2O with normal
biochemistry and microbiological analysis including cartridge-based nucleic
acid amplification test (CBNAAT) and adenosine deaminase (ADA). Noncontrast CT
brain revealed thickened, hyperdense dura along the tentorium (Fig. 3). The contrast-enhanced
magnetic resonance imaging (MRI) brain with magnetic resonance venography (MRV)
revealed engorged dural spaces, enhancement of pachymeninges, flattening of
pons, sagging of bilateral tentorial and cerebellar tonsils (Fig. 4 a-c) with
thinning of both optic nerves complicated by chronic dural venous sinus
thrombosis.
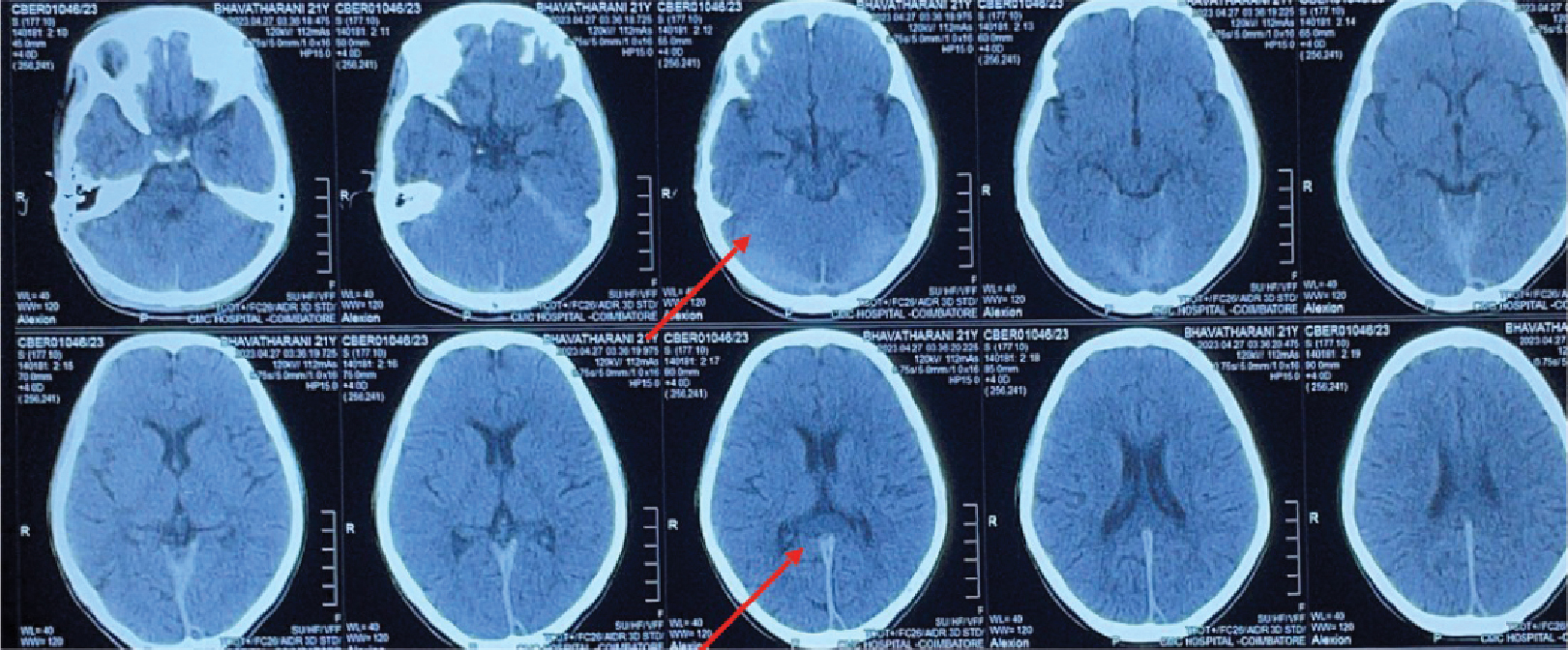
Figure 3. Noncontrast CT brain showing thickened hyperdense dural
along posterior falx and tentorium.
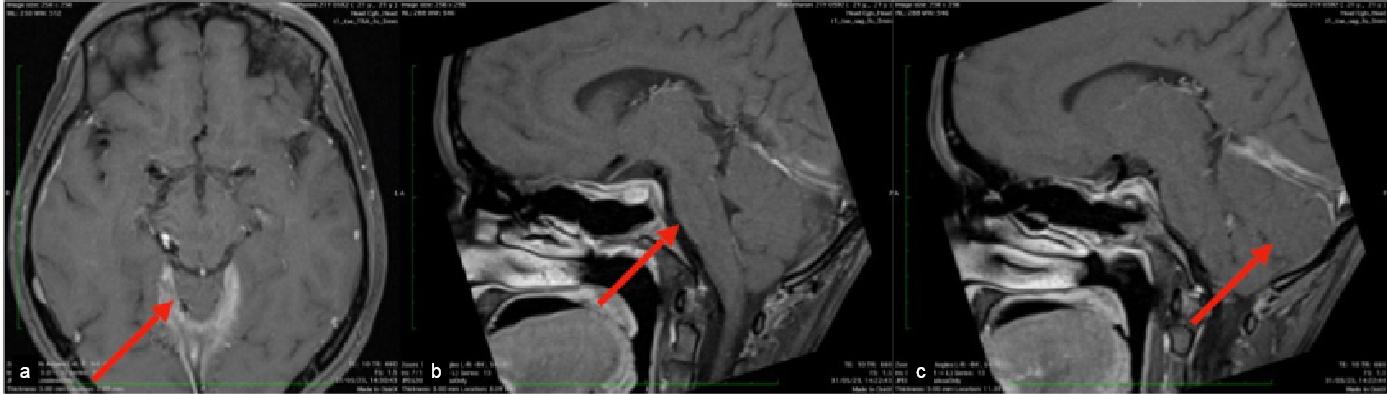
Figure 4. MRI contrast axial-engorged dural venous sinus (a);
MRI sagittal contrast flattening of pons (b);and MRI sagittal
contrast herniation of cerebellar tonsils (c).
With the background of orthostatic headache, low
CSF opening pressure, neuroimaging suggestive of engorged dural venous sinus
and sagging of brain, the probability of SIH was considered. So, a MRI
myelogram was done, which demonstrated CSF leak at left lower dorsal level
(D11-D12) (Fig. 5). A final diagnosis of SIH was made. Patient was treated with
adequate fluids, analgesics, anticoagulants, and subsequently 20 mL epidural
blood patch in the lower thoracic region. The headache and general well-being
improved and there was no further diminution of visual acuity. Repeat
neuroimaging, VEP, visual field charting was planned for during follow-up.
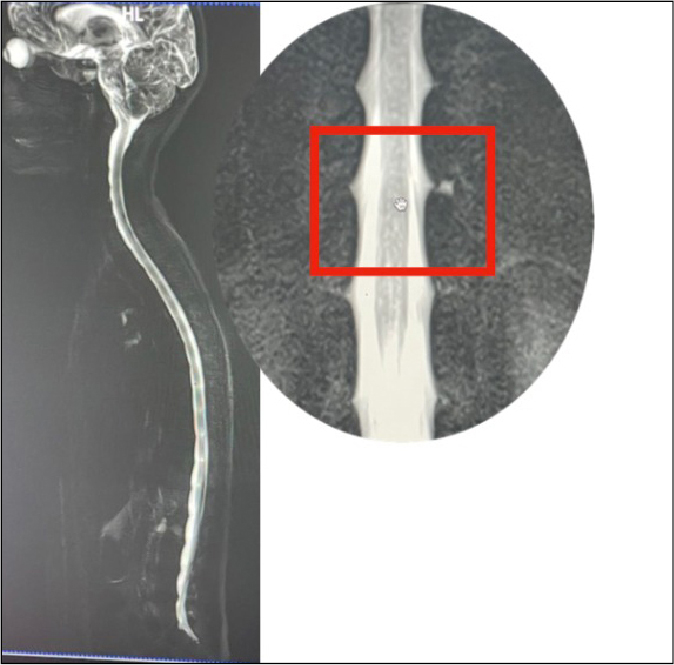
Figure 5. MRI myelography shows CSF
leak at lower dorsal level (D11-D12).
On follow-up, visual field charting showed
improvement of visual acuity 1/60 in right eye and 2/60 in left eye. VEP showed
P100 latency of 136 ms in right eye and left eye P100 latency of 120 ms with
reduced amplitude in both eyes. However, MRI showed persistence of sagging of
brain and it was planned to repeat in serial follow-ups.
On follow-up visual acuity improved in both eyes.
Right eye visual acuity - 20/200 and left eye visual acuity 20/100. Both eye
P100 latency is within limits. Neuroimaging findings are static at present.
Spontaneous intracranial hypotension is estimated
to affect 5 per 1,00,000 people per year with a female preponderance3.
It occurs when mechanical stressors (spiculated osteophytes, herniated discs,
nerve root diverticula) incite small dural tears causing CSF to leak into
extradural space4,5. It can also occur following lumbar puncture,
head trauma, spinal shunts, or rarely spontaneously. It may be exacerbated by
connective tissue disorder.
According to Monro-Kellie doctrine, skull is a
rigid compartment, which contains brain parenchyma, blood, and CSF compartment1.
CSF exerts a buoyant force suspending the cranial nerves and brain parenchyma
and prevents it from sagging downward2. These components are
balanced in a state of dynamic equilibrium in normal circumstances. When the
volume of CSF decreases in SIH, it is compensated by increase in the volume of
other components to maintain the equilibrium, which results in venous sinus
engorgement, subdural effusion, enlargement of pituitary gland, pachymeningeal
enhancement, vascular congestion of optic nerve2. This results in
traction of intracranial dura resulting in orthostatic headaches, neck pain,
and ophthalmoparesis. Similarly, downward traction of optic nerve causing
damage to the sheath of the optic nerve along with vascular congestion of the
intracranial portion of the optic nerve can account for optic neuropathy. A
case of monocular optic neuropathy associated with SIH has earlier been
reported, which was attributed to traction/compression and/or vascular
congestion of the intracranial portion of optic nerve1.
This is the first reported case of bilateral
primary optic neuropathy in a patient with SIH and this could be attributed to
downward traction of optic nerve resulting in optic nerve damage that would
eventually result in optic neuropathy.
Postural headache and optic neuropathy could be the
presenting symptoms of SIH and early recognition is of paramount
importance for preserving vision. Our case underscores the importance of early
recognition, and prompt intervention in recognizing optic neuropathy in
patients with SIH so that we can prevent the potential consequences of visual impairment
or blindness. This also emphasizes the need for the treating physician to be
vigilant in dealing with primary optic atrophy and SIH should be considered as
one of the differential diagnoses in cases with the relevant clinical
background.
Acknowledgments
We would like to extend our sincere gratitude to
Professor and Head Dr N Shobana.
1.
Goksel BK, Yildirim T, Sizmaz S, Reyhan M, Karatas
M. Optic neuropathy associated with
spontaneous intracranial hypotension. Acta Neurol Belg. 2012;112(4):361-5.
2.
Zada G, Solomon TC, Giannotta SL. A review of
ocular manifestations in intracranial hypotension. Neurosurg Focus.
2007;23(5):E8.
3.
Bond KM, Benson JC, Cutsforth-Gregory JK, Kim DK,
Diehn FE, Carr CM. Spontaneous intracranial hypotension: atypical radiologic
appearances, imaging mimickers, and clinical look-alikes. AJNR Am J
Neuroradiol. 2020;41(8):1339-47.
4.
Schievink WI. Spontaneous spinal cerebrospinal
fluid leaks and intracranial hypotension. JAMA. 2006;295(19):2286-96.
5.
HoffmannJ. Headache attributed to intracranial hypertension and hypotension. In: Mitsikostas
D, Paemeleire K (Eds.). Pharmacological Management of Headaches. Headache. Springer, Cham; 2016. https://doi.org/10.1007/978-3-319-19911-5_18